
Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
Kératodermie: causes, symptômes, diagnostic, traitement
Expert médical de l'article

Dernière revue: 07.07.2025
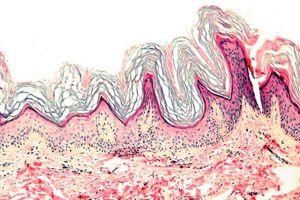
La kératodermie est un groupe de dermatoses caractérisées par une perturbation du processus de kératinisation - formation excessive de corne principalement sur les paumes et la plante des pieds.
Les causes et la pathogénèse de la maladie ne sont pas entièrement élucidées. Des recherches ont établi que les kératodermies sont causées par des mutations des gènes codant pour les kératines 6, 9 et 16. Une carence en vitamine A, des dysfonctionnements hormonaux, principalement au niveau des glandes sexuelles, ainsi que des infections bactériennes et virales jouent un rôle important dans la pathogénèse. Elles constituent l'un des symptômes de maladies héréditaires et de tumeurs des organes internes (kératodermies parapsoriasiques).
Symptômes. On distingue les kératodermies diffuses (kératodermie d'Unna-Tost, kératodermie de Meleda, kératodermie de Papillon-Lefevre, kératodermie mutilante et syndromes dont la kératodermie diffuse est l'un des principaux symptômes) et focales (kératodermie tachetée disséminée de Fischer-Buschke, acrokératoélastoïdose de Kosti, kératodermie limitée de Bruhauer-Franzesthesti, kératodermie linéaire de Fuchs, etc.).
La kératodermie de Winy-Tost (synonymes: ichtyose congénitale des paumes et des plantes des pieds, syndrome de Winy-Tost) se transmet selon le mode autosomique dominant. On observe une kératinisation excessive et diffuse de la peau des paumes et des plantes des pieds (parfois uniquement de la plante des pieds), qui se développe au cours des deux premières années de vie. Le processus pathologique cutané débute par un léger épaississement de la peau des paumes et des plantes des pieds, sous la forme d'une bande érythémateuse livide en bordure de peau saine. Avec le temps, des couches cornées lisses et jaunâtres apparaissent à leur surface. La lésion s'étend rarement au dos des poignets ou des doigts. Chez certains patients, des fissures superficielles ou profondes peuvent se former et une hyperhidrose locale est observée. Chez le patient observé par l'auteur, l'oncle maternel, le frère et le fils souffraient de kératodermie de Winy-Tost.
Des cas de lésions des ongles (épaississement), des dents et des cheveux dans la kératodermie de Winy-Tost en association avec diverses anomalies squelettiques et pathologies des organes internes, des systèmes nerveux et endocrinien sont décrits.
Histopathologie. L'examen histologique révèle une hyperkératose marquée, une granulose, une acanthose et de petits infiltrats inflammatoires dans le derme supérieur. Diagnostic différentiel. La maladie doit être différenciée des autres types de kératodermie.
La kératodermie de Meleda (synonymes: maladie de Meleda, acrokératome congénital progressif, kératose palmoplantaire transgradiente de Siemens, kératose palmoplantaire progressive héréditaire de Kogoy) est transmise selon un mode autosomique récessif. Cette forme de kératodermie se caractérise par d'épaisses couches cornées jaune-brun présentant de profondes fissures. Un liseré violet-violet de plusieurs millimètres de large est visible le long des bords de la lésion. Le processus s'étend généralement au dos des mains et des pieds, aux avant-bras et aux tibias. La plupart des patients présentent une hyperhidrose locale. À cet égard, la surface des paumes et des plantes des pieds devient légèrement humide et couverte de points noirs (canaux sudoripares).
La maladie peut se développer dès l'âge de 15 à 20 ans. Les ongles s'épaississent et se déforment.
Histopathologie. L'examen histologique révèle une hyperkératose, parfois une acanthose, et un infiltrat inflammatoire chronique du derme papillaire.
Diagnostic différentiel. La kératodermie de Melela doit être distinguée de la kératodermie d'Unna-Tost.
La kératodermie Papillon-Lefevre (synonyme: hyperkératose palmoplantaire avec parodontite) est transmise selon un mode autosomique récessif.
La maladie se manifeste entre la deuxième et la troisième année de vie. Son tableau clinique est similaire à celui de la maladie de Melela. De plus, des modifications dentaires sont caractéristiques (anomalies de l'éruption des dents de lait et des dents définitives avec développement de caries, gingivite, parodontose à progression rapide avec perte prématurée des dents).
Histopathologie. L'examen histologique révèle un épaississement de toutes les couches de l'épiderme, en particulier de la couche cornée, et des amas cellulaires insignifiants de lymphocytes et d'histiocytes dans le derme.
Diagnostic différentiel. La maladie doit être distinguée des autres kératodermies. Un signe distinctif important est la pathologie dentaire caractéristique, absente des autres formes de kératodermies diffuses héréditaires.
La kératodermie mutilante (synonymes: syndrome de Fonwinkel, kératome mutilant héréditaire) est une forme de kératodermie diffuse transmise selon le mode autosomique dominant. Elle se développe au cours de la deuxième année de vie et se caractérise par des dépôts cornés diffus sur la peau des paumes et des plantes des pieds, accompagnés d'hyperhidrose. Avec le temps, des sillons cordiformes se forment sur les doigts, entraînant des contractures et une amputation spontanée des doigts. Une kératose folliculaire se manifeste sur le dos des mains, ainsi qu'au niveau des articulations du coude et du genou. Les ongles sont altérés (souvent en verre de montre). Des cas d'hypogonadisme, d'alopécie rubis, de perte auditive et de pachyonychie ont été décrits.
Histopathologie. L'examen histologique révèle une hyperkératose sévère, une granulose, une acanthose et de petits infiltrats inflammatoires dans le derme, constitués de lymphocytes et d'histiocytes.
Diagnostic différentiel. Pour différencier la kératodermie mutilante des autres formes de kératodermie diffuse, il convient de prendre en compte en premier lieu l'effet mutilant, atypique pour les autres formes. Lors du diagnostic différentiel de toutes les formes de kératodermie diffuse, il est important de garder à l'esprit qu'elle peut être l'un des principaux symptômes de nombreux syndromes héréditaires.
Traitement. La néotigazone est indiquée dans le traitement général de la kératodermie. La dose dépend de la gravité de la maladie et est de 0,3 à 1 mg/kg de poids corporel. En l'absence de néotigazone, un traitement à long terme par vitamine A est recommandé à raison de 100 à 300 000 mg par jour. Le traitement externe consiste en l'utilisation de pommades contenant des rétinoïdes aromatiques, des kératolytiques et des stéroïdes.
 [
[ Qu'est ce qui te tracasse?
Qu'est-ce qu'il faut examiner?
Comment examiner?