
Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
Phénylpyruvine oligophrénie ou phénylcétonurie
Expert médical de l'article

Dernière revue: 05.07.2025
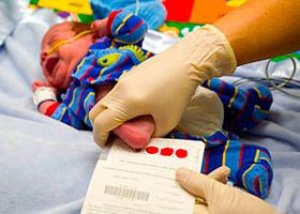
Selon les statistiques, un nouveau-né sur 10 à 15 000 est diagnostiqué avec une pathologie neurocognitive génétiquement déterminée - l'oligophrénie phénylpyruvique, qui se développe en raison d'un trouble congénital du métabolisme de l'acide aminé essentiel phénylalanine.
La maladie a été identifiée pour la première fois dans les années 1930 en Norvège par le médecin Ivar Foelling, qui l'a baptisée hyperphénylalaninémie. Aujourd'hui, cette pathologie est communément appelée phénylcétonurie, son code CIM-10 étant E70.0.
 [
[ Causes de l'oligophrénie phénylpyruvique
Les causes génétiques de l'oligophrénie phénylpyruvique sont la transmission récessive chez l'enfant de parents porteurs de deux allèles mutés de l'enzyme phénylalanine hydroxylase (phénylalanine-4-monooxygénase). De ce fait, l'enfant est totalement ou partiellement dépourvu de cette enzyme, nécessaire à la dégradation de la phénylalanine (acide α-amino-β-phénylpropionique protéinogène) en tyrosine [acide 2-amino-3-(4-hydroxyphényl)propanoïque].
Les généticiens ont établi que la pathogenèse de cette maladie est associée à plus de 500 mutations structurelles homozygotes ou hétérozygotes différentes du chromosome 12, dans les gènes de l'enzyme hépatique phénylalanine hydroxylase 12q23.2 ou 12q22-q24.1. Cette mutation génétique entraîne une diminution d'au moins 4 fois du taux de cette enzyme, nécessaire à l'oxydation de la phénylalanine contenue dans les protéines alimentaires.
Il en résulte, d'une part, une accumulation excessive de phénylalanine dans le sang, toxique pour le développement cérébral. Dans ce cas, la phénylalanine, incapable de se transformer en tyrosine, se dégrade par désamination pour produire de l'acide phénylpyruvique (acide 2-oxo-3-phénylpropionique ou phénylpyruvate). Cet acide est à son tour transformé en acide phénylacétique (phénylacétate) et en acide phényllactique (phényllactate), qui ont un effet négatif sur les cellules du cerveau et du système nerveux central.
D'autre part, les principales causes de l'oligophrénie phénylpyruvique (déficit congénital en phénylalanine hydroxylase et sa conséquence - excès de phénylalanine) entraînent une diminution des niveaux d'autres acides aminés dans le cerveau (tryptophane, thréonine, méthionine, valine, isoleucine, leucine), ce qui perturbe complètement le processus de biosynthèse des neurotransmetteurs qui transmettent l'influx nerveux - dopamine et noradrénaline - et provoque une altération progressive du développement mental et physique chez les enfants.
Symptômes de l'oligophrénie phénylpyruvique
L'excès de phénylalanine dans le corps se manifeste chez les nourrissons et ses premiers signes sont: léthargie et somnolence; problèmes de succion; convulsions; vomissements; lésions eczémateuses de l'épiderme; odeur de moisi (souris) du corps, de l'urine et de l'haleine (due à la teneur accrue en phénylacétate, qui est oxydé en phénylcétones).
Les symptômes typiques suivants de l'oligophrénie phénylpyruvique sont observés: retard de croissance; hyperkinésie musculaire, tremblements ou diminution du tonus musculaire; crises psychotiques accompagnées d'irritabilité et de colère non motivées; diminution des capacités intellectuelles ou retard mental.
Les enfants atteints de phénylcétonurie présentent également une hypopigmentation: la peau, les cheveux et les yeux sont nettement plus clairs et plus fins que ceux de leurs parents. La cause immédiate de ce symptôme est un déficit en tyrosine, qui, une fois oxydée dans les mélanocytes de l'épiderme, forme le pigment mélanine.
Comme l'ont souligné les spécialistes en pédopsychiatrie, le tableau clinique de l'oligophrénie phénylpyruvique dépend du degré de lésion des cellules cérébrales, et un bébé atteint de cette pathologie peut sembler normal durant les premiers mois de sa vie. Si la maladie n'a pas été détectée immédiatement après la naissance et que l'enfant n'a pas été traité pendant une longue période, les conséquences sont les suivantes: lésions cérébrales irréversibles et complications sous forme de retard mental grave ou profond (imbécillité ou idiotie).
Et puis les symptômes suivants de l'oligophrénie phénylpyruvique peuvent apparaître: petite taille, anomalies craniofaciales (microcéphalie, mâchoire supérieure proéminente, dents largement espacées, développement altéré de l'émail dentaire), augmentation du tonus musculaire et des réflexes tendineux, hyperactivité, mauvaise coordination des mouvements et démarche maladroite, irritabilité accrue et autres problèmes comportementaux et neurologiques pouvant aller jusqu'aux troubles mentaux.
Diagnostic de l'oligophrénie phénylpyruvique
Un diagnostic précoce de l’oligophrénie phénylpyruvique et un traitement rapide sont extrêmement importants, car ils réduisent le risque de développer un retard mental de 80-90 % à 6-8 % (selon CLIMB – National Information Centre for Metabolic Diseases, Royaume-Uni).
À cette fin, un dépistage néonatal de la phénylcétonurie doit être effectué chez les nourrissons le troisième ou le quatrième jour après la naissance (dans des circonstances exceptionnelles entre le 5e et le 8e jour) à l'aide d'un test sanguin microbiologique de la teneur en phénylalanine – appelé test de Guthrie.
Les analyses biochimiques urinaires de phénylpyruvate révèlent également une phénylcétonurie chez les nouveau-nés, mais ce test ne doit être effectué que 10 à 12 jours après la naissance. Par conséquent, la norme diagnostique internationale est la détermination de la concentration de phénylalanine dans le plasma sanguin.
Les diagnostics instrumentaux modernes, basés sur de nouvelles technologies telles que la spectrométrie de masse en tandem, permettent de détecter de nombreux troubles métaboliques congénitaux entraînant des déficiences intellectuelles. Cela permet de diagnostiquer différentiellement des pathologies métaboliques génétiques, notamment: galactosémie, cétonurie (troubles du métabolisme de la leucine, de l'isoleucine et de la valine), homocystinurie, acidurie glutarique, acidémie isovalérique, drépanocytose, tyrosinémie, hypothyroïdie congénitale, etc.
Qui contacter?
Traitement de l'oligophrénie phénylpyruvique
La diététique est pratiquement le seul traitement efficace contre l'oligophrénie phénylpyruvique, permettant ainsi au cerveau de se développer normalement. Cependant, lorsqu'elle est instaurée chez les enfants de plus de trois ou quatre ans, elle ne permet pas d'éliminer les symptômes déjà présents.
Les nourrissons présentant un déficit en phénylalanine hydroxylase ne peuvent pas consommer de lait maternel et il existe pour eux des formules spéciales sans phénylalanine: Lofenalac, Afenilak, Aminogran, Milupa pku1 (pku2 et pku3), Periflex Infanta, XP Maxamum.
Le régime exclut la consommation de viande, de poisson, de volaille, de lait, d'œufs, de fromage, de fromage blanc, de crème glacée, de légumineuses, de noix et de nombreux autres produits contenant des protéines. L'hydrolysat de caséine est utilisé comme substitut alimentaire aux produits interdits afin de maintenir l'équilibre protéique dans l'organisme. Il s'agit d'un mélange d'acides aminés sans phénylalanine extrait des protéines du lait (médicaments Berlofan ou Ipofénate).
Si les médecins pensaient auparavant que la prise de mélanges sans phénylalanine et le suivi d'un régime alimentaire particulier devaient être poursuivis pendant une période pouvant aller jusqu'à 10 ans (c'est-à-dire jusqu'à la fin des processus de myélinisation cérébrale), on sait aujourd'hui que l'arrêt du traitement peut aggraver l'oligophrénie phénylpyruvique et entraîner des conséquences irréversibles. Les patients soumis à des restrictions alimentaires ne présentent aucun symptôme.
Aujourd'hui, le médicament Kuvan, à base de dichlorhydrate de saproptérine, est un substitut synthétique du cofacteur de la phénylalanine hydroxylase tétrahydrobioptérine (BH4). Les comprimés de Kuvan, dissous dans l'eau, se prennent une fois par jour, le matin, au cours des repas. La dose est calculée individuellement en fonction du poids du patient (10 mg par kilogramme). Les effets secondaires du médicament comprennent un écoulement nasal aqueux, une congestion nasale, des maux de tête, des douleurs laryngées, des vomissements, de la diarrhée et, occasionnellement, des douleurs abdominales. La prise de ce médicament n'interrompt pas le régime anti-phénylalanine. La notice du médicament précise que son utilisation chez les enfants de moins de quatre ans n'a pas été étudiée.
Dans ce cas, le traitement de l'oligophrénie phénylpyruvique est réalisé avec une surveillance constante du taux de phénylalanine dans le sang.
L'oligophrénie phénylpyruvique, ou phénylcétonurie, est une maladie héréditaire et ne peut être prévenue. Cependant, si vous envisagez d'avoir des enfants, une prévention est possible: un test sanguin enzymatique et un test génétique permettent de déterminer si les futurs parents sont porteurs du gène mutant. Des dosages sanguins de phénylalanine peuvent également être effectués pendant la grossesse.
Comme l'écrit le Journal of Inherited Metabolic Disease, « le pronostic pour les patients atteints de cette pathologie, en l'absence de traitement, est une espérance de vie maximale pouvant aller jusqu'à 30 ans dans le statut d'une personne handicapée présentant une déficience grave de la fonction cérébrale (puisque le niveau de phénylalanine dans le sang augmentera avec le temps), et avec un traitement - jusqu'à un âge avancé, avec une éducation supérieure et une carrière réussie. »