
Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
Lissencéphalie du cerveau
Expert médical de l'article

Dernière revue: 12.07.2025
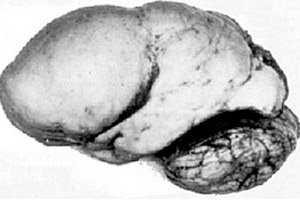
Parmi les pathologies cérébrales organiques, on distingue une anomalie congénitale du développement cérébral telle que la lissencéphalie, dont l'essence réside dans la surface presque lisse du cortex de ses hémisphères - avec un nombre insuffisant de circonvolutions et de sillons. [ 1 ]
En l'absence totale de circonvolutions, on parle d'agyrie, et la présence de plusieurs circonvolutions larges et plates est appelée pachygyrie. Ces anomalies, comme d'autres déformations réductrices du cerveau, sont codées Q04.3 dans la CIM-10.
Épidémiologie
Selon les statistiques sur les maladies rares, on compte 1 à 1,2 cas de lissencéphalie pour 100 000 nouveau-nés. [ 2 ], [ 3 ]
Selon certaines données, jusqu'à 25 à 30 % des cas de lissencéphalie classique sont observés chez les enfants atteints du syndrome de Miller-Dieker; des mutations ponctuelles et des délétions des gènes LIS1 et DCX sont détectées chez près de 85 % des patients. [ 4 ]
Des études génétiques portant sur 17 gènes associés à la lissencéphalie ont montré que la mutation ou la délétion de LIS1 représente 40 % des patients et que 23 % sont associées à la mutation DCX, suivies de TUBA1A (5 %) et de DYNC1H1 (3 %).[ 5 ]
Causes lissencéphalie
Toutes les causes connues de formation d'un cortex cérébral presque ou totalement dépourvu de circonvolutions et de sillons, qui augmentent la surface de travail du cerveau humain et assurent la productivité du système nerveux central, sont associées à des troubles du développement périnatal. Autrement dit, une lissencéphalie se développe chez le fœtus. [ 6 ]
L'échec de formation des couches du cortex cérébral du fœtus dans la lissencéphalie est le résultat d'une migration anormale des neurones qui le forment ou d'un arrêt prématuré de ce processus.
Ce processus, essentiel à l'histogenèse cérébrocorticale, se déroule en plusieurs étapes entre la 7e et la 18e semaine de grossesse. Compte tenu de sa sensibilité accrue aux mutations génétiques et à diverses influences physiques, chimiques et biologiques négatives, tout écart par rapport à la norme peut entraîner une localisation incorrecte des neurones, avec la formation possible d'une couche épaisse de matière grise du cortex, dépourvue de structure caractéristique. [ 7 ]
Dans certains cas, la lissencéphalie chez les enfants est associée aux syndromes de Miller-Dieker, de Walker-Warburg ou de Norman-Roberts.
Lire aussi – Défauts de développement du cerveau
Facteurs de risque
Outre les mutations de certains gènes, les facteurs de risque de naissance d'un enfant présentant une anomalie aussi grave comprennent le manque d'oxygène (hypoxie) du fœtus; un apport sanguin insuffisant au cerveau (hypoperfusion); un accident vasculaire cérébral aigu sous forme d'accident vasculaire cérébral périnatal; des pathologies placentaires; des infections virales de la femme enceinte (y compris TORCH); [ 8 ] des problèmes de métabolisme général et de fonction thyroïdienne; le tabagisme, l'alcool, les substances psychotropes et narcotiques; l'utilisation d'un certain nombre de médicaments; des niveaux de radiation accrus. [ 9 ]
Pathogénèse
La pathogénèse de la lissencéphalie n'est pas toujours due à des anomalies chromosomiques ou à des mutations génétiques. Cependant, certains gènes codant des protéines jouent un rôle important dans le mouvement correct des neuroblastes et des neurones le long des cellules gliales radiales, formant ainsi le cortex cérébral. Les mutations de ces gènes sont à l'origine de cette pathologie. [ 10 ]
Il s'agit notamment de mutations sporadiques (sans hérédité) du gène LIS1 sur le chromosome 17, qui régule la dynéine, protéine motrice cytoplasmique des microtubules, ainsi que du gène DCX sur le chromosome X, qui code la protéine doublecortine (lissencéphalie-X). [ 11 ]. Dans le premier cas, les spécialistes définissent une lissencéphalie classique (type I), dans le second, une lissencéphalie liée à l'X. [ 12 ]
Lorsque le gène FLN1, qui code la phosphoprotéine filamine 1, est supprimé, le processus de migration neuronale dirigée peut ne pas commencer du tout, conduisant à une absence complète de circonvolutions (agyrie). [ 13 ]
Des mutations ont été identifiées dans le gène CDK5, qui code une enzyme kinase – un catalyseur du métabolisme intracellulaire, régulant le cycle cellulaire dans les neurones du SNC et assurant leur migration normale lors de la formation prénatale des structures cérébrales.
Des modifications anormales du gène RELN sur le chromosome 7 qui provoquent des anomalies gyrales corticales dans le syndrome de Norman-Roberts entraînent une déficience de la glycoprotéine extracellulaire reelin, nécessaire à la régulation de la migration et du positionnement des cellules souches neurales pendant le développement du cortex cérébral.[ 14 ], [ 15 ], [ 16 ]
Le gène ARX code la protéine homéobox non-aristalens, un facteur de transcription qui joue un rôle important dans le cerveau antérieur et d'autres tissus.[ 17 ] Les enfants porteurs de la mutation ARX présentent d'autres symptômes, tels que des parties manquantes du cerveau (agénésie du corps calleux), des organes génitaux anormaux et une épilepsie sévère.[ 18 ],[ 19 ]
Plusieurs gènes ont été associés à la lissencéphalie. Parmi ces gènes figurent VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A et CDK5.[ 20 ]
Le cytomégalovirus (CMV) a été associé au développement d'une lissencéphalie due à une diminution de l'apport sanguin au cerveau du fœtus. La gravité de l'infection à CMV dépend de l'âge gestationnel. Une infection précoce est plus susceptible de provoquer une lissencéphalie, car la migration neuronale survient tôt dans la grossesse.[ 21 ]
De plus, le mécanisme d'apparition de cette anomalie comprend un arrêt incomplet ou tardif de la migration des neurones de la zone générative périventriculaire vers le cortex cérébral. Dans de tels cas, une lissencéphalie incomplète ou une pachygyrie se développe, caractérisée par la formation de plusieurs larges sillons et circonvolutions (la plupart étant absents).
Symptômes lissencéphalie
Les premiers signes de cette pathologie (en l'absence des syndromes mentionnés précédemment) peuvent ne pas apparaître immédiatement après la naissance, mais après un mois et demi à deux mois. Le plus souvent, les symptômes cliniques suivants sont observés:
- hypotonie musculaire, souvent associée à une paralysie spastique;
- convulsions et crises tonico-cloniques généralisées (sous forme d’opisthotonus);
- retard mental grave et retard de croissance;
- altération des fonctions neurologiques et motrices.
Les problèmes de déglutition entraînent des difficultés à nourrir le bébé. [ 22 ]
Un degré élevé de déficience neuromotrice se manifeste souvent par une tétraplégie – paralysie de tous les membres. Une déformation des mains, des doigts ou des orteils est possible.
Dans le syndrome de Norman-Roberts avec lissencéphalie de type I, des anomalies craniofaciales sont observées: microcéphalie sévère, front faiblement incliné et arête nasale large et proéminente, yeux écartés (hypertéralgie), sous-développement des mâchoires (micrognathie). [ 23 ]
Le syndrome de Miller-Dieker peut également être caractérisé par une tête anormalement petite avec un front large et haut et un nez court, des dépressions au niveau des tempes (dépressions bitemporales) et des oreilles basses et déformées.
Le syndrome de lissencéphalie sévère est caractérisé par une microcéphalie, une diminution de la taille des globes oculaires (microphtalmie), associée à une dysplasie rétinienne, une hydrocéphalie obstructive et un corps calleux absent ou hypoplasique.
Complications et conséquences
Parmi les complications de cette anomalie, les spécialistes citent une violation de la fonction de déglutition (dysphagie) et un reflux gastro-œsophagien; une épilepsie réfractaire (non contrôlée); des infections fréquentes des voies respiratoires supérieures; une pneumonie (y compris une aspiration chronique).
Les nourrissons atteints de lissencéphalie peuvent avoir des problèmes cardiaques organiques congénitaux sous la forme d'une communication interauriculaire ou d'une malformation cardiaque complexe avec cyanose (tétralogie de Fallot). [ 24 ]
Les conséquences du retard de croissance postnatal entraînent dans la plupart des cas le décès dans les 24 mois suivant la naissance.
Diagnostics lissencéphalie
Le diagnostic commence par un examen physique de l’enfant, une étude des antécédents médicaux des parents et de l’historique de la grossesse et de l’accouchement.
Pendant la gestation, un test d'ADN fœtal acellulaire, une amniocentèse ou un prélèvement de villosités choriales peuvent être nécessaires. [ 25 ] Pour plus d'informations, voir – Diagnostic prénatal des maladies congénitales
Les diagnostics instrumentaux sont utilisés pour visualiser les structures cérébrales et évaluer leurs fonctions:
- tomodensitométrie du cerveau;
- imagerie par résonance magnétique (IRM) du cerveau;
- électroencéphalogramme (EEG). [ 26 ]
Au cours de la grossesse, une lissencéphalie à l'échographie du fœtus après 20-21 semaines peut être suspectée en cas d'absence des sillons pariéto-occipitaux et calcarins et d'une anomalie de la scissure sylvienne du cerveau.
Diagnostic différentiel
Un diagnostic différentiel avec d'autres syndromes de malformations cérébrales congénitales est réalisé.
Il existe plus de 20 types de lissencéphalie, répartis en deux grandes catégories: la lissencéphalie classique (type 1) et la lissencéphalie en pavés (type 2). Chaque catégorie présente des manifestations cliniques similaires, mais des mutations génétiques différentes.[ 27 ]
L'examen cérébral de la lissencéphalie de type I révèle un cortex cérébral à quatre couches au lieu des six observées chez les patients normaux. En revanche, dans la lissencéphalie de type 2, le cortex cérébral est désorganisé et apparaît grumeleux ou nodulaire en raison d'un déplacement complet du cortex cérébral par des amas de neurones corticaux séparés par du tissu gliomesenchymateux. Les patients présentaient également des anomalies musculaires et oculaires.
- Lissencéphalie classique (type 1):
- LIS1: Lissencéphalie isolée et syndrome de Miller-Dieker (lissencéphalie associée à une dysmorphie faciale). [ 28 ]
- LISX1: Mutation du gène DCX. Comparée à la lissencéphalie causée par des mutations du gène LIS1, la DCX présente un cortex à six couches au lieu de quatre.
- Lissencéphalie isolée sans autres défauts génétiques connus
- Lissencéphalie en pavés (type 2):
- syndrome de Walker-Warburg
- Syndrome de Fukuyama
- Maladie des muscles, des yeux et du cerveau
- Les autres types ne peuvent pas être placés dans l’un des deux groupes ci-dessus:
- LIS2: syndrome de Norman-Roberts, similaire à la lissencéphalie de type I ou au syndrome de Miller-Dieker, mais sans la délétion du chromosome 17.
- LIS3
- LISX2
Microlissencéphalie: Il s'agit d'une association entre l'absence de repli cortical normal et une tête anormalement petite. Les enfants atteints de lissencéphalie régulière ont une tête de taille normale à la naissance. Les enfants dont la tête est de petite taille à la naissance sont généralement diagnostiqués avec une microlissencéphalie.
Il est également important de faire la distinction entre la lissencéphalie et la polymicrogyrie, qui sont des anomalies différentes du développement cérébral.
Qui contacter?
Traitement lissencéphalie
La lissencéphalie est un défaut organique incurable, seul un traitement symptomatique et de soutien est donc possible. [ 29 ]
Tout d'abord, il s'agit de l'utilisation d'anticonvulsivants et d'antiépileptiques, ainsi que de la pose d'une sonde gastrique (si l'enfant est incapable d'avaler seul). Le massage est utile.
Dans les cas d’hydrocéphalie sévère, le liquide céphalo-rachidien est retiré.
La prévention
Les experts recommandent aux futurs parents de consulter un conseiller génétique et aux femmes enceintes de s’inscrire auprès d’un obstétricien et d’un gynécologue en temps opportun et de se soumettre à tous les examens prévus.
Prévoir
Chez les enfants atteints de lissencéphalie, le pronostic dépend de son degré, mais le plus souvent, le développement mental de l'enfant ne dépasse pas quatre à cinq mois. Tous les enfants présentant ce diagnostic souffrent de troubles psychomoteurs sévères et d'une épilepsie difficile à traiter. [ 30 ]
Selon le NINDS (Institut national américain des troubles neurologiques et des accidents vasculaires cérébraux), l’espérance de vie maximale des personnes atteintes de lissencéphalie est d’environ 10 ans.