
Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
Néphrite héréditaire (syndrome d'Alport) chez l'enfant
Expert médical de l'article

Dernière revue: 05.07.2025
La néphrite héréditaire (syndrome d'Alport) est une glomérulopathie héréditaire non immune génétiquement déterminée, se manifestant par une hématurie (parfois avec protéinurie), un déclin progressif de la fonction rénale avec développement d'une insuffisance rénale chronique, souvent associée à une surdité neurosensorielle et à une déficience visuelle.
La maladie a été décrite pour la première fois en 1902 par LG Guthrie, qui a observé une famille où l'hématurie était présente sur plusieurs générations. En 1915, AF Hurst a décrit le développement d'une urémie chez des membres de la même famille. En 1927, A. Alport a été le premier à identifier une perte auditive chez plusieurs parents atteints d'hématurie. Dans les années 1950, des lésions oculaires liées à une maladie similaire ont été décrites. En 1972, chez des patients atteints d'hématurie héréditaire, lors d'une étude morphologique du tissu rénal, Hinglais et al. ont révélé une expansion et une stratification irrégulières des membranes basales glomérulaires. En 1985, la base génétique de la néphrite héréditaire a été identifiée: une mutation du gène du collagène de type IV (Fiengold et al., 1985).
L'étude de la nature génétique de la maladie nous a permis de conclure que les différences dans les manifestations phénotypiques de la néphrite héréditaire (avec ou sans perte auditive) sont dues au degré d'expression du gène mutant. Ainsi, à l'heure actuelle, toutes les variantes cliniques sont considérées comme des manifestations d'une seule et même maladie, et le terme « néphrite héréditaire » est synonyme de « syndrome d'Alport ».
Selon les études épidémiologiques, la néphrite héréditaire survient avec une fréquence de 17 pour 100 000 enfants.
 [
[ Causes du syndrome d'Alport
La base génétique de la maladie est une mutation du gène de la chaîne a-5 du collagène de type IV. Ce type est présent universellement dans les membranes basales du rein, de l'appareil cochléaire, de la capsule du cristallin, de la rétine et de la cornée, comme l'ont démontré des études utilisant des anticorps monoclonaux dirigés contre cette fraction du collagène. Récemment, la possibilité d'utiliser des sondes ADN pour le diagnostic prénatal de la néphrite héréditaire a été évoquée.
Il est important de tester tous les membres de la famille à l'aide de sondes ADN afin d'identifier les porteurs du gène mutant, ce qui est crucial pour le conseil médical et génétique des familles atteintes de cette maladie. Cependant, jusqu'à 20 % des familles ne comptent aucun parent atteint d'insuffisance rénale, ce qui suggère une fréquence élevée de mutations spontanées du gène anormal. La plupart des patients atteints de néphrite héréditaire ont dans leur famille des personnes atteintes d'insuffisance rénale, de perte auditive et de pathologies visuelles; les mariages consanguins entre personnes ayant un ou plusieurs ancêtres sont importants, car le mariage entre personnes apparentées augmente la probabilité de recevoir les mêmes gènes des deux parents. Des modes de transmission autosomique dominant, autosomique récessif et dominant lié à l'X ont été établis.
Chez les enfants, on distingue le plus souvent trois types de néphrite héréditaire: le syndrome d’Alport, la néphrite héréditaire sans perte auditive et l’hématurie bénigne familiale.
Le syndrome d'Alport est une néphrite héréditaire associée à une déficience auditive. Il est dû à un défaut combiné de la structure du collagène de la membrane basale glomérulaire des reins, de l'oreille et des structures oculaires. Le gène du syndrome d'Alport classique est situé au locus 21-22q du bras long du chromosome X. Dans la plupart des cas, il est transmis de manière dominante, lié au chromosome X. De ce fait, le syndrome d'Alport est plus grave chez l'homme, car chez la femme, la fonction du gène muté est compensée par un allèle sain du second chromosome, non endommagé.
La base génétique du développement de la néphrite héréditaire réside dans des mutations des gènes des chaînes alpha du collagène de type IV. Six chaînes alpha du collagène de type IV G sont connues: les gènes des chaînes a5 et a6 (Col4A5 et Col4A5) sont situés sur le bras long du chromosome X, dans la zone 21-22q; les gènes des chaînes a3 et a4 (Col4A3 et Col4A4) sont situés sur le chromosome 2; les gènes des chaînes a1 et a2 (Col4A1 et Col4A2) sont situés sur le chromosome 13.
Dans la plupart des cas (80 à 85 %), on observe un mode de transmission lié à l'X, associé à une atteinte du gène Col4A5 résultant d'une délétion, de mutations ponctuelles ou de troubles de l'épissage. Actuellement, plus de 200 mutations du gène Col4A5 ont été identifiées, responsables de l'interruption de la synthèse des chaînes a5 du collagène de type IV. Avec ce mode de transmission, la maladie se manifeste chez les enfants des deux sexes, mais elle est plus grave chez les garçons.
Les mutations des loci des gènes Col4A3 et Col4A4, responsables de la synthèse des chaînes a3 et a4 du collagène de type IV, sont transmises selon un mode autosomique. Selon les recherches, le mode autosomique dominant est observé dans 16 % des cas de néphrite héréditaire, et le mode autosomique récessif chez 6 % des patients. Une dizaine de variantes de mutations des gènes Col4A3 et Col4A4 sont connues.
Les mutations entraînent une altération des processus d'assemblage du collagène de type IV, entraînant une altération de sa structure. Le collagène de type IV est l'un des principaux composants de la membrane basale glomérulaire, de l'appareil cochléaire et du cristallin, dont la pathologie sera détectée dans le cadre d'une néphrite héréditaire.
Le collagène de type IV, qui fait partie de la membrane basale glomérulaire, est principalement constitué de deux chaînes a1 (IV) et d'une chaîne a2 (IV), ainsi que de chaînes a3, a4 et a5. Le plus souvent, dans les cas de transmission liée à l'X, la mutation du gène Col4A5 s'accompagne de l'absence des chaînes a3, a4, a5 et a6 dans la structure du collagène de type IV, et d'une augmentation du nombre de chaînes o1 et a2 dans la membrane basale glomérulaire. Le mécanisme de ce phénomène est mal connu; on suppose que la cause en est des modifications post-transcriptionnelles de l'ARNm.
L'absence des chaînes a3, a4 et a5 dans la structure du collagène de type IV des membranes basales glomérulaires entraîne leur amincissement et leur fragilité aux premiers stades du syndrome d'Alport, qui se manifeste cliniquement le plus souvent par une hématurie (plus rarement par une hématurie avec protéinurie ou une protéinurie seule), une perte auditive et un lenticone. La progression de la maladie entraîne un épaississement et une altération de la perméabilité des membranes basales aux stades avancés, avec prolifération des collagènes de types V et VI, se traduisant par une augmentation de la protéinurie et une diminution de la fonction rénale.
La nature de la mutation à l'origine de la néphrite héréditaire détermine en grande partie sa manifestation phénotypique. En cas de délétion du chromosome X associée à une mutation simultanée des gènes Col4A5 et Col4A6, responsables de la synthèse des chaînes a5 et a6 du collagène de type IV, le syndrome d'Alport est associé à une léiomyomatose de l'œsophage et des organes génitaux. Selon les données de recherche, en cas de mutation du gène Col4A5 associée à une délétion, on observe une gravité plus importante du processus pathologique, une association de lésions rénales avec manifestations extrarénales et le développement précoce d'une insuffisance rénale chronique, par rapport à une mutation ponctuelle de ce gène.
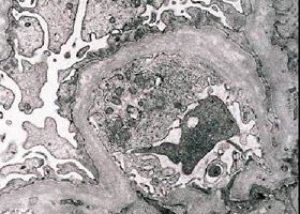
Morphologiquement, la microscopie électronique révèle un amincissement et une stratification des membranes basales glomérulaires (en particulier la lamina densa) ainsi que la présence de granules denses aux électrons. Les lésions glomérulaires peuvent être hétérogènes chez un même patient, allant de lésions mésangiales focales minimes à la glomérulosclérose. La glomérulite du syndrome d'Alport est toujours immunonégative, ce qui la distingue de la glomérulonéphrite. Les signes caractéristiques comprennent le développement d'une atrophie tubulaire, une infiltration lymphohistiocytaire et la présence de « cellules spumeuses » contenant des inclusions lipidiques (lipophages). À mesure que la maladie progresse, un épaississement et une destruction prononcée des membranes basales glomérulaires sont révélés.
Certaines modifications du système immunitaire sont mises en évidence. Les patients atteints de néphrite héréditaire présentent un taux d'IgA diminué et une tendance à l'augmentation des concentrations d'IgM dans le sang. Le taux d'IgG peut augmenter aux premiers stades de la maladie et diminuer aux stades ultérieurs. L'augmentation des concentrations d'IgM et d'IgG pourrait être une réaction compensatoire en réponse au déficit en IgA.
L'activité fonctionnelle du système lymphocytaire T est réduite; une diminution sélective des lymphocytes B responsables de la synthèse d'Ig A est notée, le lien phagocytaire de l'immunité est perturbé, principalement en raison de la perturbation des processus de chimiotaxie et de digestion intracellulaire dans les neutrophiles
Lors de l'examen d'une biopsie rénale chez des patients atteints du syndrome d'Alport, les données de microscopie électronique révèlent des modifications ultrastructurales de la membrane basale glomérulaire: amincissement, rupture de structure et division de la membrane basale glomérulaire, avec modification de son épaisseur et irrégularité des contours. Aux premiers stades de la néphrite héréditaire, ce défaut entraîne l'amincissement et la fragilité de la membrane basale glomérulaire.
L'amincissement des membranes glomérulaires est un signe plus favorable et plus fréquent chez les filles. Un signe microscopique électronique plus constant dans la néphrite héréditaire est la rupture de la membrane basale, dont la gravité est corrélée à la gravité du processus.
Symptômes du syndrome d'Alport chez les enfants
Les premiers symptômes du syndrome d'Alport, sous forme de syndrome urinaire isolé, sont le plus souvent détectés chez les enfants de moins de trois ans. Dans la plupart des cas, la maladie est détectée par hasard. Le syndrome urinaire est détecté lors d'un examen préventif de l'enfant, avant son admission en crèche ou lors d'une infection urinaire aiguë (IVA). En cas de pathologie urinaire lors d'une IVA, la néphrite héréditaire, contrairement à la glomérulonéphrite acquise, ne présente pas de période de latence.
Au stade initial de la maladie, la santé de l'enfant est peu affectée, la persistance et la résistance du syndrome urinaire étant caractéristiques. L'un des principaux signes est une hématurie de gravité variable, observée dans 100 % des cas. Une augmentation du degré d'hématurie est observée pendant ou après des infections respiratoires, une activité physique ou après des vaccinations préventives. Dans la plupart des cas, la protéinurie ne dépasse pas 1 g/jour. Au début de la maladie, elle peut être instable, puis augmente avec la progression de la maladie. Une leucocyturie à prédominance lymphocytaire peut être présente périodiquement dans le sédiment urinaire, ce qui est associé au développement de modifications interstitielles.
Par la suite, la fonction rénale partielle est altérée et l'état général du patient se dégrade: intoxication, faiblesse musculaire, hypotension artérielle, souvent une déficience auditive (surtout chez les garçons) et parfois une déficience visuelle apparaissent. L'intoxication se manifeste par une pâleur, une fatigue et des maux de tête. Au stade initial de la maladie, la perte auditive n'est généralement détectée que par audiographie. La perte auditive associée au syndrome d'Alport peut survenir à différents moments de l'enfance, mais le plus souvent, elle est diagnostiquée entre 6 et 10 ans. Chez l'enfant, la perte auditive débute par les hautes fréquences, atteignant un degré significatif en conduction aérienne et osseuse, passant d'une perte auditive de conduction à une perte auditive de perception. La perte auditive peut être l'un des premiers symptômes de la maladie et précéder le syndrome urinaire.
Dans 20 % des cas, les patients atteints du syndrome d'Alport présentent des modifications des organes visuels. Les anomalies les plus fréquemment détectées concernent le cristallin: sphérophokie, lenticone antérieur, postérieur ou mixte, et diverses cataractes. Dans les familles atteintes du syndrome d'Alport, la myopie est fréquente. Plusieurs chercheurs observent régulièrement des modifications périmaculaires bilatérales dans ces familles, sous la forme de granulations blanchâtres ou jaunâtres brillantes dans le corps jaune. Ils considèrent ce signe comme un symptôme constant ayant une valeur diagnostique importante dans le syndrome d'Alport. KS Chugh et al. (1993), dans une étude ophtalmologique, ont observé chez les patients atteints du syndrome d'Alport une baisse de l'acuité visuelle dans 66,7 % des cas, un lenticone antérieur dans 37,8 %, des taches rétiniennes dans 22,2 %, une cataracte dans 20 % et un kératocône dans 6,7 %.
Chez certains enfants atteints de néphrite héréditaire, notamment en cas d'insuffisance rénale, on observe un retard important du développement physique. À mesure que l'insuffisance rénale progresse, une hypertension artérielle se développe. Chez les enfants, elle est plus souvent détectée à l'adolescence et chez les personnes plus âgées.
Les patients atteints de néphrite héréditaire se caractérisent par la présence de divers stigmates (plus de 5 à 7) de dysmorphogénèse du tissu conjonctif. Parmi ces stigmates, les plus fréquents sont l'hypertélorisme oculaire, le palais haut, les anomalies de l'occlusion, la forme anormale des oreillettes, la courbure de l'auriculaire et l'espace entre les doigts et les pieds. La néphrite héréditaire se caractérise par l'uniformité des stigmates de dysmorphogénèse au sein d'une même famille, ainsi que par leur fréquence élevée parmi les parents des sujets dont la maladie est transmise.
Aux premiers stades de la maladie, on observe une diminution isolée des fonctions rénales partielles: transport des acides aminés, électrolytes, fonction de concentration, acidogénèse. Les modifications ultérieures affectent l'état fonctionnel des parties proximale et distale du néphron et se caractérisent par des troubles partiels combinés. Une diminution de la filtration glomérulaire survient plus tard, plus souvent à l'adolescence. À mesure que la néphrite héréditaire progresse, une anémie se développe.
Ainsi, la néphrite héréditaire se caractérise par une évolution par étapes: d'abord, un stade latent ou des symptômes cliniques cachés, se manifestant par des modifications minimes du syndrome urinaire, puis une décompensation progressive du processus se produit avec une diminution de la fonction rénale et des symptômes cliniques manifestes (intoxication, asthénie, retard de développement, anémie). Les symptômes cliniques apparaissent généralement quelle que soit la stratification de la réaction inflammatoire.
La néphrite héréditaire peut se manifester à différentes périodes d'âge, ce qui dépend de l'action du gène, qui est dans un état réprimé jusqu'à un certain moment.
Classification
Il existe trois types de néphrite héréditaire
- Option I – se manifeste cliniquement par une néphrite avec hématurie, perte auditive et lésions oculaires. L'évolution de la néphrite est progressive et évolue vers une insuffisance rénale chronique. Le mode de transmission est dominant, lié au chromosome X. Morphologiquement, une altération de la structure de la membrane basale, son amincissement et sa division sont mis en évidence.
- Option II – se manifeste cliniquement par une néphrite avec hématurie sans perte auditive. L'évolution de la néphrite est progressive et évolue vers une insuffisance rénale chronique. Le mode de transmission est dominant, lié au chromosome X. Morphologiquement, un amincissement de la membrane basale des capillaires glomérulaires (en particulier laminadensa) est observé.
- Option III: hématurie familiale bénigne. L'évolution est favorable, sans insuffisance rénale chronique. Le mode de transmission est autosomique dominant ou autosomique récessif. Dans le cas d'une transmission autosomique récessive, l'évolution de la maladie est plus grave chez les femmes.
Diagnostic du syndrome d'Alport
Les critères suivants sont proposés:
- la présence d’au moins deux patients atteints de néphropathie dans chaque famille;
- l'hématurie comme principal symptôme de néphropathie chez le proposant;
- la présence d’une perte auditive chez au moins un membre de la famille;
- développement d'une insuffisance rénale chronique chez un ou plusieurs parents.
Dans le diagnostic de diverses maladies héréditaires et congénitales, une grande importance est accordée à une approche globale de l'examen et, surtout, à l'attention portée aux données obtenues lors de l'établissement de l'arbre généalogique de l'enfant. Le diagnostic du syndrome d'Alport est considéré comme valide lorsque 3 des 4 signes typiques sont détectés chez le patient: présence d'hématurie et d'insuffisance rénale chronique dans la famille, présence d'une surdité neurosensorielle, pathologie visuelle, et détection de signes de clivage de la membrane basale glomérulaire avec modification de son épaisseur et irrégularité des contours lors de l'examen microscopique électronique de la biopsie.
L'examen du patient doit inclure des méthodes de recherche clinique et génétique; une étude ciblée de l'anamnèse; et un examen général du patient prenant en compte les critères diagnostiques pertinents. Au stade de compensation, la pathologie ne peut être détectée qu'en se concentrant sur des syndromes tels qu'une charge héréditaire, une hypotension, de multiples stigmates de dysembryogenèse et des modifications du syndrome urinaire. Au stade de décompensation, des symptômes extrarénaux peuvent apparaître, tels qu'une intoxication grave, une asthénie, un retard de développement physique et une anémie, qui se manifestent et s'intensifient avec une diminution progressive de la fonction rénale. Chez la plupart des patients présentant une diminution de la fonction rénale, on observe: une diminution de l'acidogénèse et de l'aminogénèse; 50 % des patients notent une diminution significative de la fonction sécrétoire rénale; une amplitude limitée des fluctuations de la densité optique urinaire; une perturbation du rythme de filtration, puis une diminution de la filtration glomérulaire. Le stade d'insuffisance rénale chronique est diagnostiqué lorsque les patients présentent un taux élevé d'urée dans le sérum sanguin (plus de 0,35 g/l) pendant 3 à 6 mois ou plus, et une diminution de la filtration glomérulaire à 25 % de la norme.
Le diagnostic différentiel de la néphrite héréditaire doit être réalisé principalement en cas de forme hématurique de glomérulonéphrite acquise. La glomérulonéphrite acquise présente le plus souvent un début aigu, 2 à 3 semaines après l'infection, des signes extrarénaux, notamment une hypertension dès les premiers jours (au contraire, une hypotension dans la néphrite héréditaire), une diminution de la filtration glomérulaire au début de la maladie, et l'absence d'altération des fonctions tubulaires partielles, alors que dans la néphrite héréditaire, elles sont présentes. La glomérulonéphrite acquise se manifeste par une hématurie et une protéinurie plus prononcées, ainsi que par une augmentation de la VS. Les modifications typiques de la membrane basale glomérulaire, caractéristiques de la néphrite héréditaire, ont une valeur diagnostique.
Le diagnostic différentiel de la néphropathie dysmétabolique est réalisé en cas d'insuffisance rénale chronique, de maladies rénales hétérogènes cliniquement diagnostiquées dans la famille, et il peut exister un spectre de néphropathies allant de la pyélonéphrite à la lithiase urinaire. Les enfants se plaignent souvent de douleurs abdominales et, périodiquement, lors de la miction, de sédiments urinaires (oxalates).
Si une néphrite héréditaire est suspectée, le patient doit être adressé à un service spécialisé de néphrologie pour clarifier le diagnostic.
Qu'est-ce qu'il faut examiner?
Comment examiner?
Quels tests sont nécessaires?
Qui contacter?
Traitement du syndrome d'Alport
Le traitement comprend des restrictions sur les efforts physiques intenses et l'exposition à l'air libre. Le régime alimentaire est complet, avec des apports suffisants en protéines complètes, lipides et glucides, en tenant compte de la fonction rénale. La détection et le traitement des foyers d'infection chroniques sont essentiels. Les médicaments suivants sont utilisés: ATP, cocarboxylase, pyridoxine (jusqu'à 50 mg/jour), chlorure de carnitine. Les cures sont administrées 2 à 3 fois par an. En cas d'hématurie, des plantes médicinales sont prescrites: ortie, jus d'aronia, achillée millefeuille.
Des publications nationales et étrangères font état de traitements par prednisolone et de l'utilisation de cytostatiques. Cependant, il est difficile d'en évaluer l'effet.
En cas d’insuffisance rénale chronique, on a recours à l’hémodialyse et à la transplantation rénale.
Il n'existe pas de traitement spécifique (pathogénique efficace) de la néphrite héréditaire. Toutes les mesures thérapeutiques visent à prévenir et à ralentir le déclin de la fonction rénale.
L'alimentation doit être équilibrée et riche en calories, en tenant compte de l'état fonctionnel des reins. En l'absence de troubles fonctionnels, l'alimentation de l'enfant doit contenir suffisamment de protéines, de lipides et de glucides. En présence de signes de dysfonctionnement rénal, les apports en protéines, glucides, calcium et phosphore doivent être limités, ce qui retarde le développement d'une insuffisance rénale chronique.
L’activité physique doit être limitée; il est conseillé aux enfants d’éviter le sport.
Il convient d'éviter tout contact avec des patients contagieux et de réduire le risque de développer une maladie respiratoire aiguë. L'assainissement des foyers d'infection chronique est nécessaire. Les enfants atteints de néphrite héréditaire ne sont pas vaccinés à titre préventif; la vaccination n'est possible que pour des raisons épidémiologiques.
Les traitements hormonaux et immunosuppresseurs sont inefficaces dans la néphrite héréditaire. Certains effets positifs (réduction de la protéinurie et ralentissement de la progression de la maladie) sont observés lors d'une utilisation prolongée et pluriannuelle de ciclosporine A et d'inhibiteurs de l'ECA.
Dans le traitement des patients, des médicaments sont utilisés pour améliorer le métabolisme:
- pyridoxine - 2-3 mg/kg/jour en 3 prises pendant 4 semaines;
- cocarboxylase - 50 mg par voie intramusculaire tous les deux jours, soit un total de 10 à 15 injections;
- ATP - 1 ml par voie intramusculaire tous les deux jours, 10 à 15 injections;
- vitamine A - 1000 UI/an/jour en 1 prise pendant 2 semaines;
- Vitamine E - 1 mg/kg/jour en 1 prise pendant 2 semaines.
Ce type de thérapie permet d'améliorer l'état général des patients, de réduire les dysfonctionnements tubulaires et est réalisé en cures 3 fois par an.
Le lévamisole peut être utilisé comme immunomodulateur - 2 mg/kg/jour 2 à 3 fois par semaine avec des pauses entre les doses de 3 à 4 jours.
Selon les données de recherche, l’oxygénation hyperbare a un effet positif sur la gravité de l’hématurie et du dysfonctionnement rénal.
La méthode la plus efficace pour traiter la néphrite héréditaire est la transplantation rénale rapide. Dans ce cas, il n'y a pas de récidive de la maladie chez le greffé; dans un faible pourcentage de cas (environ 5 %), une néphrite peut se développer dans le rein transplanté, associée à des antigènes de la membrane basale glomérulaire.
Le diagnostic prénatal et la thérapie par génie génétique constituent des pistes prometteuses. Les expériences animales montrent une grande efficacité du transfert de gènes normaux responsables de la synthèse des chaînes alpha du collagène de type IV dans le tissu rénal, permettant ainsi la synthèse de structures collagènes normales.
Prévision
Le pronostic de la néphrite héréditaire est toujours grave.
Les critères pronostiques défavorables pour l'évolution de la néphrite héréditaire sont:
- genre masculin;
- développement précoce d’une insuffisance rénale chronique chez les membres de la famille;
- protéinurie (plus de 1 g/jour);
- épaississement des membranes basales glomérulaires selon la microscopie;
- névrite acoustique;
- suppression dans le gène Col4A5.
Le pronostic de l’hématurie familiale bénigne est plus favorable.